
The journey from a foundational laboratory discovery to a transformative patient therapy is arduous, particularly in the realm of rare genetic diseases. For decades, these conditions—often caused by a single, specific error in the vast human genome—served as poignant illustrations of medical need unmet by therapeutic possibility. The advent of CRISPR-Cas9 gene editing, dubbed Gene Editing 1.0, ignited a revolution by providing a programmable system to cut DNA at targeted locations. However, its translation for direct in vivo therapy for many inherited disorders faced significant hurdles: its reliance on creating double-strand DNA breaks (DSBs) introduced risks of uncontrolled insertions, deletions, and chromosomal rearrangements. The evolution to Gene Editing 2.0—encompassing base editing, prime editing, and highly refined delivery technologies—marks a pivotal transition from blunt cutting to precise molecular rewriting, offering a new paradigm for curing, rather than merely managing, rare monogenic diseases.
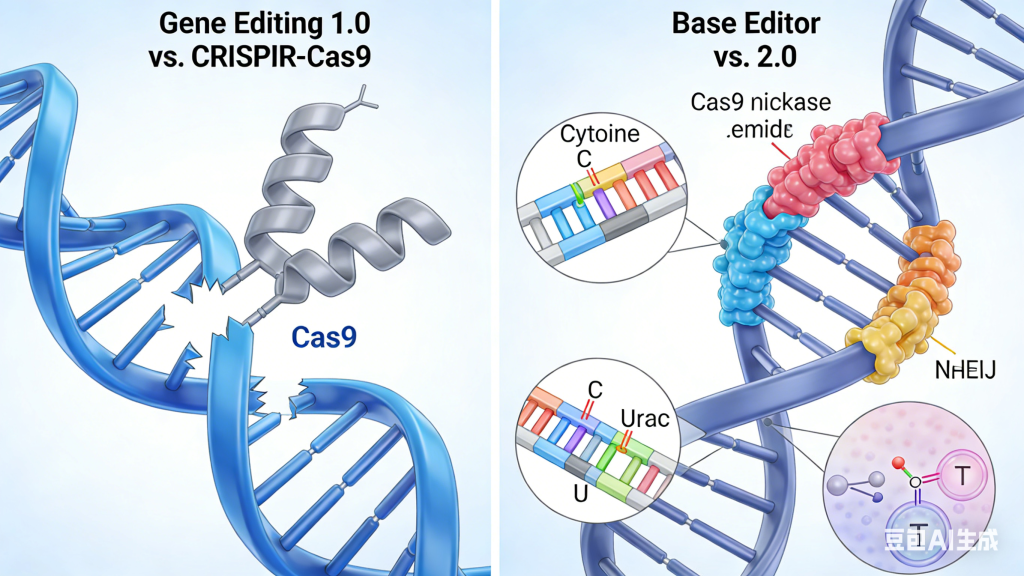
Gene Editing 2.0 technologies are defined by their enhanced precision and minimized collateral damage. Base editors, first described by David Liu’s team, function as programmable chemical converters. Without cutting the DNA backbone, they directly and irreversibly convert one DNA base pair into another (e.g., C•G to T•A, or A•T to G•C). This allows for the direct correction of point mutations, which constitute the majority of known pathogenic variants in rare diseases like sickle cell anemia (caused by an A•T to T•A transversion in the HBB gene) or certain forms of progeria. By avoiding DSBs, base editors dramatically reduce the generation of indel byproducts, enhancing safety.
For more complex corrections beyond simple point mutations, prime editing, a subsequent breakthrough, acts as a “search-and-replace” genomic word processor. A prime editor uses an engineered reverse transcriptase fused to a Cas9 nickase, guided by a prime editing guide RNA (pegRNA) that specifies both the target site and contains the desired new genetic sequence. It nicks one DNA strand, uses the pegRNA as a template to write the corrected sequence directly into the genome, and then leverages cellular repair mechanisms to incorporate the edit. This system can mediate targeted insertions, deletions, and all 12 possible base-to-base conversions, theoretically addressing nearly 90% of known pathogenic human genetic variants, including those responsible for diseases like Tay-Sachs or cystic fibrosis, with exceptional fidelity.
The clinical translation of these tools hinges on parallel breakthroughs in delivery. The in vivo treatment of somatic cells, especially in hard-to-reach tissues like neurons or muscle, requires vectors of unprecedented specificity and efficiency. Next-generation adeno-associated virus (AAV) capsids, engineered through directed evolution for enhanced tropism (e.g., crossing the blood-brain barrier), and non-viral delivery systems such as lipid nanoparticles (LNPs) targeted to specific cell types, are critical for making systemic, tissue-specific editing a clinical reality. A therapeutic regimen for a neuromuscular disorder like Duchenne Muscular Dystrophy, for instance, now envisions a one-time intravenous infusion of LNPs carrying a base editor designed to permanently correct a premature stop codon in the dystrophin gene within muscle satellite cells.
Early clinical pipelines reflect this shift. While ex vivo CRISPR-Cas9 editing for sickle cell disease represents the vanguard of Editing 1.0, the first wave of in vivo Editing 2.0 trials is now targeting the liver. Programs are underway for diseases like hereditary transthyretin amyloidosis (hATTR), where a single base editor is delivered to hepatocytes via LNP to permanently inactivate the mutant TTR gene. The central nervous system is the next frontier, with preclinical studies demonstrating the potential of dual-AAV systems to deliver compact base editors into the brain to correct mutations in genes like MECP2 for Rett syndrome or CLN3 for Batten disease.
Despite the promise, significant translational challenges persist. Off-target editing, though reduced, must be characterized with exquisite sensitivity using advanced assays like GUIDE-seq or CIRCLE-seq. Immune responses to bacterial-derived editors or delivery vectors require careful management. Furthermore, the ethical and regulatory landscape for permanent somatic editing is complex, demanding robust long-term follow-up frameworks to monitor for delayed effects. The scientific community emphasizes a measured, evidence-based pace, prioritizing conditions with clear genotype-phenotype correlations and high unmet need.
In conclusion, Gene Editing 2.0 represents more than an incremental improvement; it signifies a maturation of the field from conceptual genome cleavage to practical genome surgery. By offering a suite of tools capable of making precise, predictable corrections without relying on error-prone DNA break repair, these technologies are dismantling the previously insurmountable barrier between identifying a genetic cause and implementing a definitive cure. The path from laboratory to bedside for rare genetic diseases is being fundamentally shortened and rerouted, pointing toward a future where a one-time, curative genomic intervention becomes a tangible reality for thousands of patients living with once-untreatable conditions.




Discuss